炎症是关节疼痛的“幕后元凶”:揭秘骨关节炎的发病机制
2025-07-27 22:55:37
长久以来,骨关节炎(Osteoarthritis, OA)常被人们简单地归因于关节的“老化”或“磨损”。中老年人膝盖上下楼梯时的隐痛、手指关节的僵硬变形,似乎都是岁月流逝无可避免的代价。然而,现代医学研究正在不断揭示一个更为复杂且活跃的真相:骨关节炎并非一场寂静的衰退过程,而是一场从单纯的机械磨损逐步升级为显著炎症风暴的慢性病变过程。正是这场病变过程中持续存在的“炎症”,成为了患者关节疼痛难以熄灭的根源。

郭锐 副主任医师
昆山市第三人民医院副主任医师
硕士研究生
2004年从事骨科工作,先后赴上海长海医院、江苏大学附属医院、日本三重大学附属医院、苏州大学附属第二医院进修学习,积累了丰富的临床经验,对骨科常见病和多发病能准确的诊断和治疗
并参与《踝关节骨折后下胫腓前韧带损伤对踝关节稳定作用的研究》、《胸腰椎爆裂骨折椎管内难复性骨块的应对策略》 、《脊柱手术中骶棘肌的保护及术后功能锻炼防止腰背部衰弱综合症》等科研项目
在国内核心期刊、外文期刊发表多篇论文
骨关节炎的发展历程
第一阶段:磨损的序曲——机械退变与微损伤
故事的开端,确实与“磨损”相关,但这磨损并非均匀平静。这是生物力学改变以及机械性微损伤所造成的。此阶段疼痛的特点是可能轻微、间歇性(如久坐后站起、长时间行走后),休息可缓解。影像学(X片)可显示关节间隙轻微变窄(软骨变薄)、早期骨赘形成。
● 软骨:首当其冲的“减震垫”。关节软骨光滑坚韧,覆盖在骨端,是天然的减震器和润滑层。它的健康依赖于软骨细胞(数量稀少但功能关键)精心维护的细胞外基质——主要由胶原纤维网和蛋白聚糖构成。年复一年的使用、反复的冲击负荷、关节的轻微不稳(如韧带旧伤)、甚至体重增加带来的额外压力,都在软骨上留下痕迹。胶原纤维网络开始出现微小断裂,蛋白聚糖逐渐流失。面对损伤,软骨细胞最初试图修复,合成更多的基质成分。但它们的修复能力本就有限。持续的异常应力会干扰软骨细胞的正常功能,甚至导致部分细胞过早死亡(凋亡)。最终,修复与分解的平衡被打破,分解代谢逐渐占据上风。
● 软骨下骨:悄然改变的“地基”。软骨下方的骨质并非被动支撑。在OA早期,可作出对上方软骨损伤和局部应力改变的适应性反应。承受异常压力的区域,骨密度增加、变硬,试图提供更强支撑。而应力较低或存在微小骨折的区域,可能形成充满液体或纤维组织的囊性变。这部分区域的破骨细胞(拆骨)和成骨细胞(建骨)的活动失衡,最终导致骨小梁结构紊乱。
● 骨赘(骨刺):并非“罪魁”而是“维稳尝试”。关节边缘或韧带附着处常出现新骨增生,形成骨赘。这本质上是机体试图增加关节接触面积、分担负荷、重新获得稳定的一种代偿反应。虽然骨赘可能刺激周围组织引起疼痛,但它本身是结果而非OA启动的主因。
第二阶段:炎症信号发出——滑膜炎症的登场
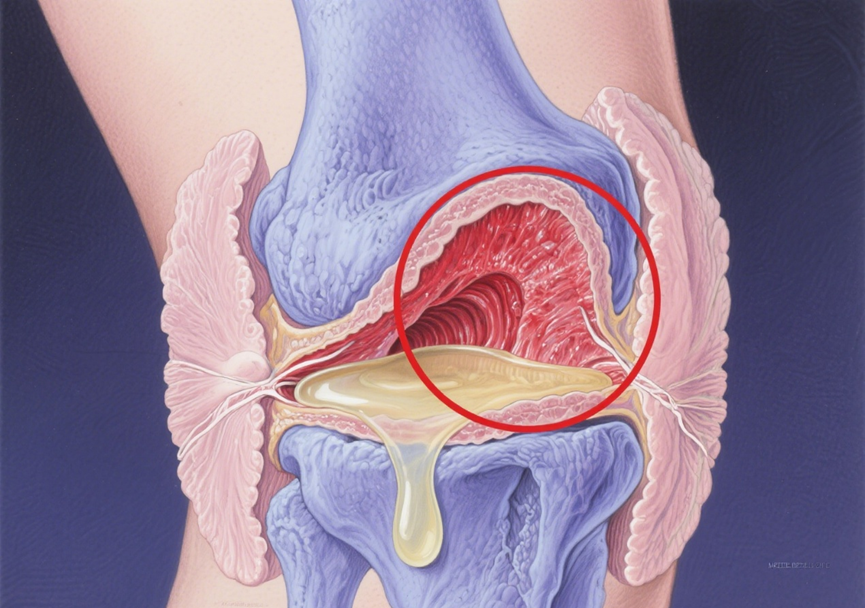
随着关节内环境的持续恶化,一个关键角色被激活:滑膜及其伴随的低度慢性炎症成为这个阶段的主角。这是OA从“单纯退变”转向“炎症性关节病”的关键转折点,也是疼痛加剧的核心环节。主要是由损伤相关分子模式(DAMPs)驱动的慢性炎症(滑膜炎)。
此阶段疼痛的特点是变得更频繁、更明显,可能出现静息痛(不动也痛)、夜间痛、关节僵硬感(尤其是晨僵,虽然通常短于类风湿关节炎)。关节可出现肿胀(滑膜增厚和积液)、局部皮温升高。影像学上关节间隙变窄更明显,骨赘增多增大。
磨损脱落的软骨碎片(如胶原、蛋白聚糖片段)、微小的晶体(如钙盐)、甚至受损或死亡细胞释放的内容物(如ATP、HMGB1蛋白),这些本应被封闭在组织内部的物质,现在散落到关节腔。它们被免疫系统识别为“危险信号”——DAMPs。滑膜组织中富含巨噬细胞,它们是关节的“哨兵”。当DAMPs被巨噬细胞表面的模式识别受体捕获,巨噬细胞瞬间被激活。被激活的巨噬细胞和滑膜成纤维细胞,开始大量分泌强效的促炎因子,例如白细胞介素-1β (IL-1β)、肿瘤坏死因子-α (TNF-α)、白细胞介素-6 (IL-6)等。持续的促炎因子刺激导致滑膜组织增生、血管增多、炎性细胞浸润(巨噬细胞、淋巴细胞为主),形成滑膜炎。发炎的滑膜本身变得肥厚、充血,成为炎症因子持续产生的源头。滑膜炎症还会导致关节液(滑液)分泌增多、性质改变(变得稀薄、炎症因子浓度升高),进一步损害关节内环境。
第三阶段:疼痛的交响——炎症是主旋律
关节疼痛并非单一原因所致,而是一个复杂的“交响乐团”。在这个乐团中,炎症因子扮演着指挥和主奏的角色。
● 直接刺激痛觉神经末梢:炎症因子(如IL-1β, TNF-α, PGE2, 缓激肽, 神经生长因子NGF)能直接作用于分布在关节囊、韧带、滑膜、软骨下骨甚至部分半月板边缘的痛觉感受器(伤害性感受器),降低其激活阈值,使其对原本无害的刺激(如正常活动、轻微压力)变得敏感,产生疼痛信号。
● 神经长入与疼痛传递:OA晚期,受损的软骨下骨区域甚至退变的软骨边缘,可能出现异常的神经纤维长入(神经支配异常)。同时,炎症环境促进NGF等神经营养因子释放,这些因子不仅促进神经生长,还会使已有的痛觉神经变得更加敏感,对炎症因子和机械刺激的反应更剧烈。
● 大脑处理疼痛变得“敏感”:持续的外周炎症和疼痛信号,会“上传”至脊髓和大脑。在中枢神经系统层面,负责处理和传递疼痛信号的神经元也会发生可塑性改变,变得更加兴奋,放大疼痛信号,甚至在没有新的外周刺激时也产生疼痛。这解释了为什么一些OA患者即使关节结构破坏并不特别严重,疼痛却异常剧烈和顽固。
● 软骨下骨是关键疼痛源:软骨下骨的硬化、囊变、骨髓水肿(MRI上可见)以及异常的神经长入,使其成为OA疼痛的重要来源,尤其在负重时。骨髓水肿区域压力增高,刺激骨内敏感的神经末梢。疼痛导致活动减少,肌肉(尤其是关节周围的稳定肌)萎缩、力量减弱。肌肉无力又使得关节稳定性下降,异常负荷增加,进一步加剧软骨磨损和炎症,形成恶性循环。肌肉本身的功能失调和可能的触发点也会产生疼痛。
因此,OA发展到中后期的疼痛,其主旋律是炎症因子驱动的外周和中枢神经敏感化。单纯的“磨损”理论无法解释为何休息时也痛、为何疼痛程度常与影像学严重程度不完全匹配。而炎症,正是连接关节组织破坏与患者主观痛苦体验的核心桥梁。
骨关节炎的防治策略
明白了疼痛的关键在于“磨损”基础上的“炎症”和“神经敏感”,治疗策略就很清晰了:“减磨”的同时,还有侧重“抗炎”,结构严重破坏时需手术。
(1)基础治疗(贯穿始终,侧重“减磨”):
● 患者教育与自我管理:主要目的在于理解疾病本质,建立合理预期。
● 减重:减轻膝关节、髋关节等负重关节的机械负荷。
● 运动疗法:科学运动能改善疼痛、功能,甚至可能轻度延缓结构进展。包括低冲击有氧运动(游泳、骑车)增强心肺功能;力量训练(尤其关节周围肌群)。提高关节稳定性、分担软骨负荷;柔韧性训练改善关节活动度。
● 物理疗法:用于辅助缓解症状、改善功能。包括热疗、冷疗、电疗、按摩、支具/矫形器(如膝OA的楔形鞋垫、护膝)等。
(2)药物治疗(侧重“抗炎”):
● 外用非甾体抗炎药(NSAIDs):首选,直接作用于局部,副作用小,有效缓解轻中度疼痛和炎症。
● 口服NSAIDs:用于中重度疼痛,需严格评估胃肠道、心血管、肾脏风险,选择最低有效剂量、最短疗程。
● 关节腔内注射:
①糖皮质激素可强效抗炎,短期(数周至数月)缓解中重度疼痛和肿胀(尤其有滑膜炎时),但不宜频繁使用(可能加速软骨破坏)。
②透明质酸(黏弹性补充治疗)可改善滑液润滑和减震功能,对部分患者有效,起效相对慢但持续时间可能较长。
③其他如对乙酰氨基酚(止痛效果弱于NSAIDs,且无抗炎作用);度洛西汀(抗抑郁药,被批准用于慢性OA疼痛,可能作用于中枢敏感化)等。
(3)手术治疗(结构严重破坏时):
● 关节镜下清理术:作用有限且短暂,适应证需严格把握。
● 截骨术:矫正关节力线异常(如膝内/外翻),适合相对年轻、活动量大、病变主要累及单侧间室的特定患者。
● 人工关节置换术:终极解决方案,用于终末期OA,效果显著,能极大改善疼痛和功能,提高生活质量。
总结
通过本文可认识到炎症在疼痛中的核心作用,不仅解释了患者为何深陷疼痛之苦,更为精准干预提供了方向。既要减轻关节负担、延缓磨损,更要积极控制关节内的慢性炎症。
对于深受骨关节炎困扰的人们而言,通过科学管理体重、坚持合理运动、在医生指导下适时使用抗炎药物、甚至在必要时接受成熟的手术治疗,完全有可能有效控制炎症之火,显著减轻疼痛。
告别“单纯磨损”的旧识,拥抱“控制炎燃”的新途,让每一次行走不再沉重,让生命的活力在关节间重新自由流淌。
 CHTV 百姓健康微信
CHTV 百姓健康微信